| 生物制品境外转移到境内的三大注册路径 | 您所在的位置:网站首页 › 沙特FDA注册 要提供外箱设计 › 生物制品境外转移到境内的三大注册路径 |
生物制品境外转移到境内的三大注册路径
|
来源:雪球App,作者: 研发客,(https://xueqiu.com/4376152234/204193343) 路径一:用境外生产的产品供应境内首次上市; 路径二:技术转移后的产品用于我国首次上市; 路径三:技术转移后的产品用于中国境内首次上市。 报告人| 左珺 和铂医药药政法规事务部副总裁 整理| 研发客 过往,当谈到治疗用生物制品境外技术转移到境内,申请人一般都比较谨慎,原因在于监管方面对生物制品有一些特别要求。随着国家药监局加入ICH,从2018年到2020年,ICH各项指导原则在我国逐步落地实施,加之监管科学和“以患者为中心”理念的推进,中国药监正与国际接轨。
胰岛素能否成为境外转境内的模版? 当前,化药从境外转移到境内生产的案例较多,但生物制品的案例鲜有发生。今年6月,《已上市生物制品药学变更研究技术指导原则》《生物制品变更受理审查指南》等出台后,生物制品由境外技术转移到境内的注册路径和技术要求也相对明晰了。 笔者现列举成功获批的由境外转移到境内生产的胰岛素产品。
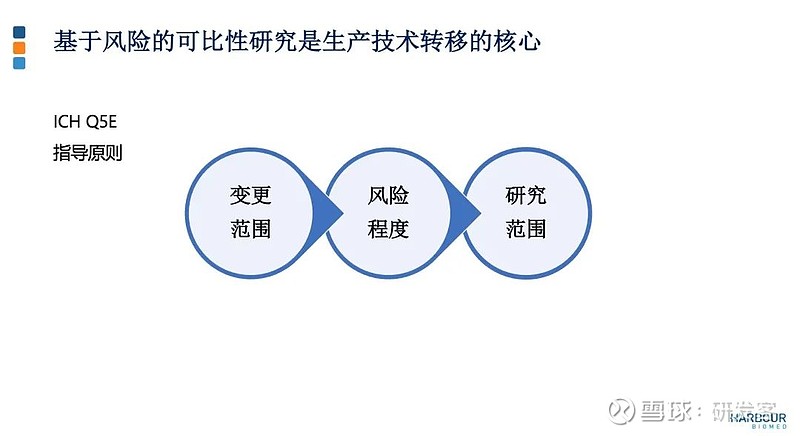
如上图所示,在诺和诺德的胰岛素产品线中,有7个成功转移到境内生产,礼来的胰岛素产品赖脯胰岛素注射液也于今年获得批准。这些产品境外技术转移到境内生产的模式均是进口原液,在境内灌装制剂。 这些胰岛素产品成功转移到境内生产的经验和模式,能否用于其他生物制品?基于对生物制品管理的模式,这类产品技术转移到境内生产的模式还不能普遍用于其它生物制品,特别是单抗类的产品。原因在于: 首先,对分子量很大、结构复杂的生物制品,不允许原液和制剂的跨境分段生产。生物制品生产技术转到境内时,不可进口原液,在境内灌装制剂。胰岛素产品本身是特例,因为胰岛素与一般的生物制品不一样,其分子结构确定,分子量小,在历史上按化药监管,后转为生物制品。其次,原液和制剂在境内的分段生产是有条件的。在同一质量管理体系下,原液和制剂可以分段生产,如果不在一个质量管理体系下,分段生产一般不被允许。 生产场地技术转移的成功因素 生产场地技术转移是否成功,基于风险的可比性研究至关重要。有关可比性研究的指南可以参考ICH Q5E和国家药监局发布的相关指导原则。通常情况下,生产场地的技术转移会伴随生产设备和工艺变更等产生的变化。
因此 ,在进行可比性研究时,要基于变更类型及变更引发的风险等级来制定可比性研究范围。基于产品可比性研究结果判断场地技术转移前后的产品质量是否等同或高度相似。 对于质量属性的差异,基于对产品知识了解和经验,预判这些差异会否对产品有效性和安全性产生影响。
不同变更导致不同的风险等级。如果不涉及生产用细胞系变更或给药系统变更,这里列举的其它类变更属于中风险或低风险。对于生产技术转移过程中伴随的低风险或中风险的变更,可比性研究可基于 CMC的分析可比性,如果必要再加上非临床研究的可比性,证明生产场地变更前后产品的质量是否可比。 另外,生物制品的风险等级与研发阶段高度关联。在Ⅲ期临床试验阶段,生产场地和生产工艺基本锁定,如果发生场地变更或工艺变更,风险相对较高。 产品能否尽早顺利获批,对每家公司至关重要。在此阶段,要尽可能避免生产场地或生产工艺的重大变更。
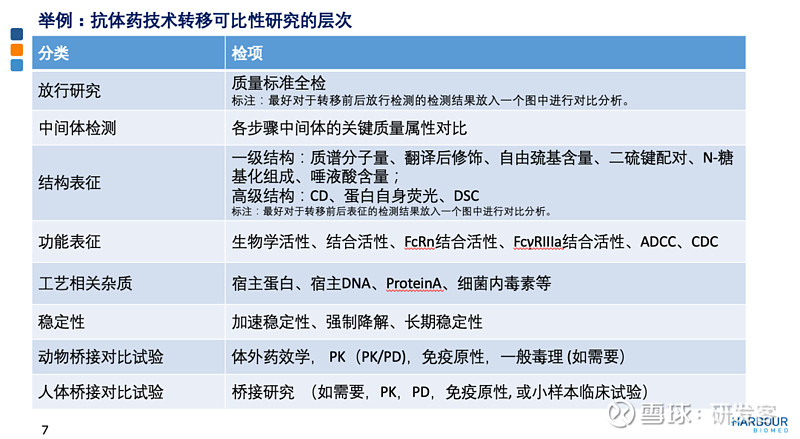
以抗体产品为例来说明技术转移中可比性研究的层次。首先,基于对产品了解和关键质量属性、质量控制策略的把控上,进行CMC层面头对头研究,变更包括前后放行检测、中间体检测、表征分析、杂质谱、稳定性研究加上工艺比对,以此判断生产场地转移前后产品质量是否可比。 基于风险管理的三种变更路径 如果需要,基于风险,还要完成非临床层面的可比性研究,需要与转移前的样品进行组间对照。如果CMC和非临床研究不能得出产品质量可比,就需要对临床研究进行比对。
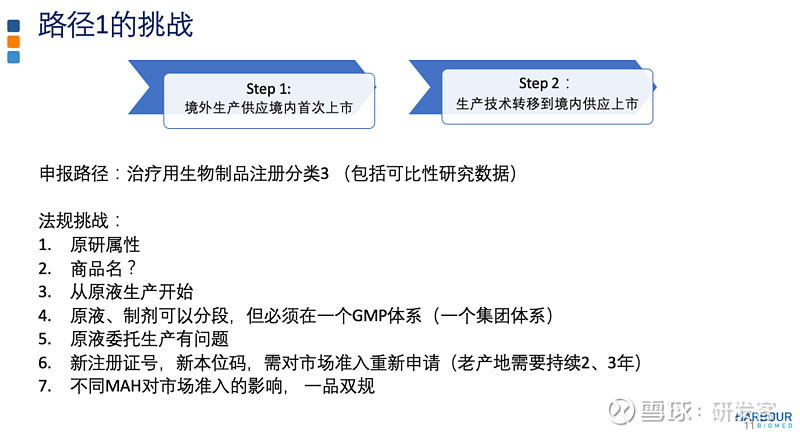
对于生物制品由境外转移到境内,有哪些注册路径可以考虑? 如上图所示,路径1较传统,用境外生产的产品供应境内首次上市。在现有法规下,如果做好计划和执行,中国参与境外临床研究,以境外生产的产品供应境内外市场,可以实现境内外同步递交上市申请。在临床试验过程中,进行技术转移到境内并完成可比性研究。基于申请人与CDE的沟通交流,按照药品上市注册申请的程序和要求提出申请,获得批准后就可用境内生产的产品供应中国市场,实现由境外生产供应转境内生产供应。为避免生产技术转移可能出现的临床研究要求,这一阶段不建议涉及生产用细胞系的变更或给药系统的变更。
目前还没有除胰岛素产品以外的其它生物制品生产场地转移案例,这里列举的案例是境外产品上市后的生产场地转移所做的可比性研究。这个重组糖蛋白产品上市后,涉及原液生产场地从欧洲变更到美国,同时有细胞培养工艺的改进、纯化工艺调整。企业基于 CMC分析的可比性,还有非临床可比性研究,证明了生产场地的转移是成功的。场地转移后,产品与转移前的质量可比,最后也获得了FDA的批准。
路径下的法规挑战包括原液制剂分段生产、商品名、市场准入等问题。
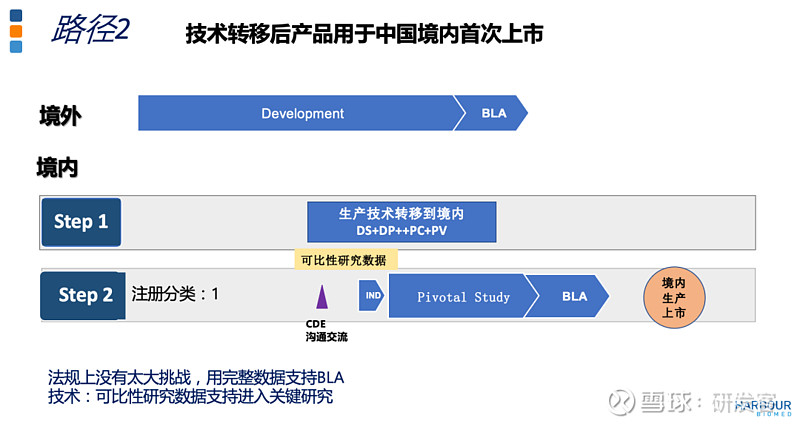
路径2是技术转移后的产品用于我国首次上市。这一路径要求生产技术转移要在产品研发的早期进行。基于可比性研究,与CDE沟通,是否能免除早期临床研究,如果必要可用PK研究桥接。这个路径下,在我国进行关键性临床研究,一定要用技术转移后的产品完成,用完整的研究数据支持生物制品上市,由此可使用境内生产的产品在我国的首次上市。
这里列举的是一个单抗产品在开展临床III期前的生产场地技术转移,包括原液和制剂,原液还涉及培养基变更及工艺调整和优化。这不是严格的由境外转到境内的变更。用于支持场地变更的数据包括:变更前后工艺的比对,变更前后原研和制剂三批次的分析记录、放行检测、原液和制剂的稳定性研究,中间体杂质谱的检测以及表征分析,最后基于可比性研究结果,CDE批准了产地变更。
路径3也是技术转移完成后的产品用于中国境内首次上市。例如,一个生物制品早期在境外开发,由国内一家企业license in,在中国境内进行开发。技术转移到境内后,完成可比性研究。这个转移和开发与路径2基本相同,支持上市的关键性临床研究的样品需要由技术转移到境内生产的产品完成。通过可比性研究,可以利用境外早期数据,或者PK研究来桥接境外早期数据。 展望未来 对境外生产技术转移到境内,首先从技术考量上,基于风险的可比性研究是核心,针对场地技术转移伴随的变更类型和风险评估制定可比性研究策略。对产品知识、工艺、关键质量属性详细深入的了解是可比性研究成功的关键;其次,要与监管部门尽早沟通,针对生产场地技术转移需完成的可比性研究策略达成一致。行业正处在新的法规体系下,新法规意味着新机会,有关生物制品由境外技术转移到境内,需要申请人和监管机构共同探索、积累经验。期盼有更多生物制品成功转移到境内,惠及中国患者。
生物制品上市后药学研究变更研讨会主讲嘉宾 互动环节 主持人: 闫小军 百济神州药政事务部负责人,高级副总裁 讨论嘉宾: 杨萌 罗氏药学技术法规政策中国负责人 李宝泉 信达生物注册于知识产权部注册副总监 左珺 和铂医药副总裁,注册事务部负责人 吴正宇 诺和诺德注册事务总监 何海琼 百济神州药政事务部高级注册总监 问:对近期颁布的《已上市生物制品药学变更研究技术指导原则(试行)》(以下简称《生物制品变更技术指导原则》,大家有哪些印象最深的部分? 答:《生物制品变更技术指导原则》,是专门针对生物制品出台的药学变更指导原则,历经多年反复讨论和修订,新指导原则最终于今年6月发布,业界人心鼓舞。我们能从中感受到药审中心(CDE)付出了很多心血,也读出了监管方与行业充分交流的开放心态。新指导原则的前言和基本考量部分尤为出彩,最大的亮点在于明确鼓励申请人使用先进的技术工具和管理方法,确定更为合理的变更分类,凸显指导原则的前瞻性,强调药品全生命周期的药学变更管理理念。 变更研究的技术要求基于产品有效性、安全性和质量可控予以细化和规范化,充分参考了世界卫生组织(WHO)、FDA及EMA的有关指导原则,在理念和细则上既与先进的国际监管实践与行业经验接轨,又充分考虑了中国特有的监管制度。 《生物制品变更技术指导原则》是对变更管理的“升级”,申请人实施基于风险、基于产品和工艺知识的生物制品药学变更管理时,若在某一领域经验不足,则会遇到巨大挑战。而这样的挑战,同样是监管机构需要面对的。其中,“如何控制风险”是企业和监管部门一直面对的老问题。如何既给予工业界灵活度,又确保不产生额外监管风险或使风险可控,确实需要智慧。 国家药监局和省药监局都负责审查和判定申请人提出的生物制品药学变更分类,而国/省之间、各省之间的尺度有可能不一致。新的法规制度和指导原则下,如何达成国/省、省/省统一理解及执行是关键。新指导原则的良好实施尚有许多需要推进之处,例如,省药监局对指导原则规定的技术要求(尤其是中等和微小变更)如何理解和实施,业界期待尽快清晰化、规范化和统一化。 问:在MAH制度下,国内有不少跨省委托生产的成功案例。MAH和生产场地跨省及两个生产厂跨省对产品的制造或检验的管理,有何经验可分享? 答:若两个生产厂跨省,同一变更在不同省给出的处理方式有可能不一样。例如,涉及两个生产厂分别位于A省和B省,如果发生中等变更,在向MAH所在地报告的同时也涉及A省和B省的属地监管,MAH所在地省局有可能不接受该变更的备案/报告,例如,省药监局认为涉及核准的工艺和标准(制检规程)须由CDE批准或咨询CDE,不接受备案;若该MAH在生产厂所在省外的第三个省,则情况会更加复杂。建立规范化机制,各省药监局和CDE对上市后变更的分类界定和监管尽量达成一致,可以帮助企业更有效地管理上市后药学变更,确保合规和病人用药。 多点生产包括两种情况,一是在同一生产场地多点选择,二是不同生产地址多点选择,如跨省生产。监管机构要求用一套制造检定规程对多点生产的药品进行监管,以保证多点生产的质量一致。现实情况下, 因为生产设备,厂房设计等差异,不同生产厂很难按照同一个制检规程生产同一个产品,但企业在从一个生产厂向另一个生产厂进行技术转移时,会展开综合的可比性研究,确保尽管存在设备、工艺和/或检验方法流程的差异,多点生产的产品最终质量是可比的。因此,企业需要在实施多点生产前进行细致的差距分析和相应的风险评估,并依据风险设定可比性研究方案,确保技术转移后的产品与原生产厂的产品质量可比。必要时,需与监管部门进行沟通交流。监管机构在核准制检规程时,在确保多点生产产品质量可比的前提下,应允许每个生产厂的制检规程基于生产实际而存在不同。 当前的变更备案是“公示”的,任何人都可查阅公示信息。这就使得申请人处在两难境地,若公示描述的变更事项不充分,则无法得到有关监管部门和市场准入相关方的认可;若公示描述清晰细致,则会过度暴露商业秘密。而无论备案类变更还是报告类变更,在跨省多点生产下,可能涉及多个省局的监管和信息披露,环节越多,企业商业秘密发生泄露的担忧越大, 监管部门是否可以考虑适当调整公示内容。 国外生物制药公司的分段/多点生产是常态,国内公司会逐步从国内走向国际,进行分段和多点生产,变更过程中要面对动态和复杂的变化,更需要监管部门在现有体制下对多点和分段生产的支持。因此,在管理生物药品生命周期中的药学变更时,应分层次和分阶段进行。采用多条生产线和多个生产场地时,企业要在生产场地变更,临床研究和商业化对接上进行系统考量,企业的变更策略要与监管部门共同探讨,以避免对药物开发的进程带来不必要的阻碍。 问:在同一MAH制度下可批准多个产地,是否DS(原料药/原液)DP(制剂)分段在不同厂生产同样可行? 答:国家已经逐渐开放生物制品的多点生产,但尚未开放分段生产,因为生物制品的风险程度更高,监管方面会更加谨慎。欧美日等国外监管机构把责任放在MAH上,法律体制和监管已较为成熟,他们的GMP监管和实施基础扎实。可供借鉴的是,在美国FDA,会特别关注血液制品和疫苗等高风险产品的分段生产,并给予特别的监管措施,例如FDA对血液制品的分段生产采取专门的“合作生产”措施予以监管,多个生产企业分段生产血液制品时,每个生产企业都获得BLA且在工艺变更时将作为整体考虑,上下游一变,整个流程都变,各家获得BLA的生产企业可能都要对变更进行研究并采取行动,相当于形成了合作生产联盟。 监管不能只针对具有成熟的质量管理体系的公司,也不能只迁就质量管理水平低下的公司,而要兼顾所有类型企业。并且,应当鼓励和发展具有更高质量管理水平的公司采用更为灵活的生产运营布局。 问:说到分段生产,CDE允许在同一GMP体系分段生产,您能举一个案例么? 答:从既有案例看,若是境外生产的生物制品,可接受其在境外生产厂分段生产。因其境外的生产监管不纳入生产许可制度,默认其上市监管首先由境外监管机构承担并出具监管的官方凭据,甚至其变更需以境外监管机构的批准为在境内注册申报的条件。在分段生产环节,对于进口产品的监管会借助于国外监管机构的监管,研发生产的模式与产品生产者所在地的监管制度和工业界实操相匹配。 但若是境内生产的生物制品,生产监管和注册管理的“双轨监管”都由境内管辖并实施许可制度,在药品上市“双轨监管”下的监管承担方,若不能确保其监管风险受控,则不敢放开分段生产,因而限制了生物制品平台化专业分工的发展。例如,原液生产在药明康德上海工厂,制剂生产在药明康德苏州工厂,在符合特定前提条件下是能够被接受的。“必须在同一质量管理体系下”是最为关键的前提条件,如果在不同质量管理体系下分段生产,目前还不被允许。 问:制剂生产中是否需要明确几批DS来合批成一批DP,如果不同,是否需要进行合批和验证? 答:关于原液混批的审评和监管要求有些波折,但在2020年时已经较为清晰合理。原液混批可参考《中国药典》四部的“人用重组DNA蛋白制品总论”和最新修订的GMP生物制品附录,按要求开展DS批次混合为半成品以制备DP,并提供相应的“混合工艺”的验证资料。在实际的审评案例中,往往被要求提供经验证的最大混合批数,但这一要求的必要性尚待商榷,在各批次均为合格品的情况下,逻辑上不可能混合后变成“不合格”,从国外监管实践看并不要求验证最大混合批数,当然,建立可接受标准的合理性时需要对此有所考虑。 尽管业界往往并不认为混合有必要验证其“混合工艺”,但囿于《中国药典》规定如此,若混合工艺的验证成为一道“必答题”,则需结合实际生产情况与CDE洽谈具体要求。例如审评中希望将其规定为最多只能合三批,但若制剂批量远大于原液批量,确需多于三批原液才足够填装制剂生产设备一个批量的经验证容积,则应当放开最大混合批数,不可能空空放着制剂的大容量罐,却每批都只填入罐底的浅浅一层。至于多个尾批任意混合等极端情况,监管方要求企业尽量减少混合批数,则建议企业与监管方充分协商(混合批数越多潜在担忧越大),应尽量依据科学做出监管决策。 问:多批原液混合工艺的验证有没有具体要求?比如,直接做放行检测就可以么?是否需要做稳定性研究?还是需要做多批次或者有可比性的研究? 答:这个问题可以考虑和监管机构沟通探讨。验证方案的设计是要为验证的目的服务的。如果在工艺中引入原液混批,基于风险,就可比性而言,一般是不需要进行综合的(extensive)可比性研究的。 问:混批并没有写到变更指导原则中,请问这属于国家药监局还是省药监局的监管范畴? 答:鉴于其列入GMP附录,且不改变关键的工艺和标准,应属于GMP范畴,而不应在注册提交的级别。当具体事项不清晰时,申请人可先找省药监局反映,若省药监局同意备案,现有备案流程非常快;若省局明确不属于省局的监管范畴(例如省药监局认为涉及核准的制检规程,须由CDE批准),则要按审批类变更提交给CDE。 问:I期试验直接用原液灌装,II期或III期样品经过配方工艺(formulation)研制出的DP需要开展什么研究?除了药学还需要动物或PK桥接试验吗? 答:如I期试验样品,需要有临床前研究支持,除了按照《创新药临床药理学研究技术指导原则(征求意见稿)》,不但要做传统PK,还要观察受试者的安全性和有效性。在临床试验期间发生的变更,尤其是关键临床研究前和上市后变更,考虑重点不一样。I到II期也是如此逻辑。到了II期、III期使用配方工艺,III期临床研究后的变更,理论上要开展完整的可比性研究,在早期,很多时候只能做比对研究。 生物制品变更系列其他文章 基于风险的生物制品上市后变更管理 五步骤完成单抗药生产规模扩大变更 * 感谢左珺博士、杨萌博士、海琼博士、吴正宇博士对本文的指导和斧正,并授权使用PPT。感谢杨森公司张宁、泰格医药周晴瑶、常建青、诺和诺德公司吴正宇、罗氏公司杨萌、陈宇参与讨论,感谢DIA中国、泰格医药联合研发客组织举办此次沙龙。 总第1466期 访问研发客网站可浏览更多文章 网页链接
|









【本文地址】